
- Capvaxive – A 21-Valent Pneumococcal Conjugate Vaccine
- An Epinephrine Nasal Spray (neffy) for Anaphylaxis
- Vonoprazan (Voquezna) for Nonerosive GERD
- A New RSV Vaccine (mResvia) for Adults ≥60 Years Old
- In Brief: New Warning for Fezolinetant (Veozah)
- Figure 1: Pneumococcal Vaccine Recommendations for Adults 19-64 Years Old (online only)
- Figure 2: Pneumococcal Vaccine Recommendations for Adults ≥65 Years Old (online only)
- Addendum: Effectiveness of mRNA COVID-19 Vaccines (online only)
- In Brief: Afamitresgene Autoleucel (Tecelra) for Synovial Sarcoma (online only)
- In Brief: Erzofri — Another Once-Monthly Paliperidone Formulation (online only)
- Mark Abramowicz, M.D., President has disclosed no relevant financial relationships.
- Jean-Marie Pflomm, Pharm.D., Editor in Chief has disclosed no relevant financial relationships.
- Cynthia Covey has disclosed no relevant financial relationships.
- Review the efficacy and safety of Capvaxive, a 21-valent pneumococcal conjugate vaccine, for prevention of invasive pneumococcal disease and pneumococcal pneumonia in adults.
- Description: A 21-valent pneumococcal conjugate vaccine (PCV21).
- Indication: Prevention of invasive pneumococcal disease and pneumococcal pneumonia in adults.
- Coverage: PCV21 includes serotypes that collectively account for 85% of IPD cases in adults ≥65 years old.
- Adverse Effects: Injection-site reactions, fatigue, headache, myalgia, and arthralgia.
- Dosage: Single 0.5-mL dose given by intramuscular injection.
- Cost: One dose costs $287.
- Conclusion: The CDC Advisory Committee on Immunization Practices (ACIP) recommends PCV21 as an option for adults when a PCV vaccine is recommended.
Outline
Tables |
The FDA has licensed Capvaxive (PCV21; Merck), a 21-valent pneumococcal conjugate vaccine, for prevention of invasive pneumococcal disease (IPD) and pneumococcal pneumonia in adults. Four other pneumococcal vaccines are currently available in the US: Prevnar 20 (PCV20), Vaxneuvance (PCV15), and Prevnar 13 (PCV13) are conjugate vaccines licensed for use in persons ≥6 weeks old, and Pneumovax 23 (PPSV23) is a pneumococcal polysaccharide vaccine licensed for use in persons ≥2 years old (see Table 1).1

PNEUMOCOCCAL DISEASE ― Streptococcus pneumoniae is a common cause of bacterial pneumonia. Adults ≥65 years old and persons with certain medical and immunocompromising conditions (see Figure 1) are at increased risk of developing IPD. In the US, vaccination against S. pneumoniae has substantially reduced the incidence of pneumococcal disease in children and adults.
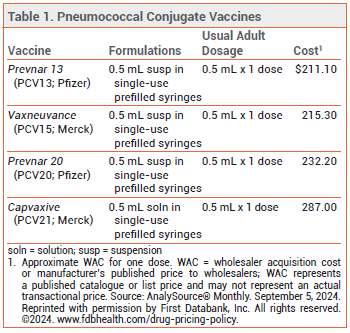
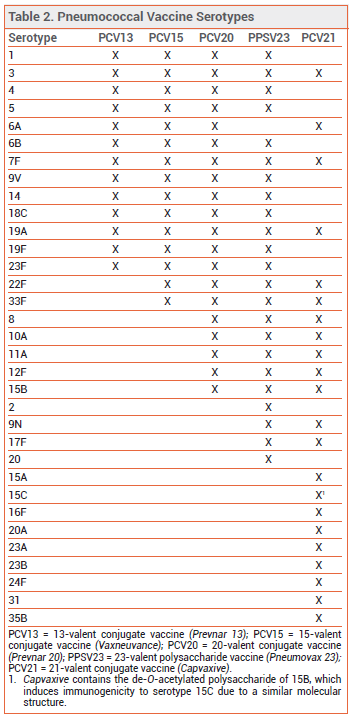
THE NEW VACCINE ― Like PCV13, PCV15, and PCV20, PCV21 contains capsular polysaccharide antigens conjugated to a protein carrier (nontoxic diphtheria CRM197 toxin). The 21 serotypes included in Capvaxive account for 85% of IPD cases in adults ≥65 years old; PCV20 includes serotypes that account for 54% of these cases. PCV21 includes 8 serotypes that are not included in any of the other currently available pneumococcal vaccines (see Table 2); these serotypes account for 30% of IPD cases in adults ≥65 years old. Some serotypes included in the other vaccines were excluded from PCV21 because they are no longer commonly associated with pneumococcal disease in adults.
Capvaxive does not protect against serotype 4, which is included in other pneumococcal vaccines and is responsible for ≥30% of IPD cases in certain populations (i.e., adults <65 years old living in some western US regions who are homeless, have chronic lung disease or alcohol use disorder, smoke, or use injectable drugs).
CLINICAL STUDIES ― FDA approval of Capvaxive was based on the results of 3 trials (STRIDE-3, STRIDE-5, and STRIDE-6; STRIDE-5 is unpublished) evaluating its immunogenicity in both vaccine-naive and previously vaccinated adults. In STRIDE-3 and STRIDE-6, immune responses elicited by PCV21 were noninferior to those elicited by PCV15, PCV20, and PPSV23 for all shared serotypes.2,3
In STRIDE-5 (summarized in the package insert), 1080 adults ≥50 years old were randomized to receive PCV21 with or 30 days after receiving a quadrivalent influenza vaccine. Immune responses elicited when the two vaccines were administered together were noninferior to those elicited when PCV21 was given 30 days after influenza vaccination for 20 of 21 pneumococcal serotypes (not serotype 23B) and for 3 of 4 influenza strains.
ADVERSE EFFECTS ― Injection-site reactions (pain, swelling, and erythema), fatigue, headache, myalgia, and arthralgia were the most common adverse effects reported with Capvaxive in clinical trials. Adverse effects were similar when Capvaxive was administered alone or with a quadrivalent inactivated influenza vaccine. Like other conjugate vaccines, Capvaxive is contraindicated for use in persons with a severe allergy to diphtheria toxoid.
PREGNANCY ― The American College of Obstetricians and Gynecologists (ACOG) recommends pneumococcal vaccination for pregnant women at increased risk of severe pneumococcal disease.4 No adequate data are available on the use of PCV21 in pregnant women. No adverse developmental outcomes were observed in animal studies.
ACIP RECOMMENDATIONS ― The CDC Advisory Committee on Immunization Practices (ACIP) recommends PCV21 as an option for adults who are candidates for pneumococcal vaccination.5 Previously unvaccinated adults who are ≥65 years old or 19-64 years old with certain underlying medical conditions or other risk factors should receive a one-time dose of PCV21 or PCV20, or a dose of PCV15 followed ≥1 year later by PPSV23.
Specific recommendations for pneumococcal vaccination of adults based on vaccination status, age, and risk factors are available in Figures 1 and 2.


DOSAGE AND ADMINISTRATION ― Capvaxive is supplied in single-dose, prefilled syringes that should be stored in a refrigerator. It should be given intramuscularly as a single 0.5-mL dose.
CONCLUSION ― The new 21-valent pneumococcal conjugate vaccine Capvaxive contains serotypes that collectively account for 85% of invasive pneumococcal disease in older adults in the US, including 8 serotypes that are not included in other currently available pneumococcal vaccines. It is a recommended option for adults who are candidates for pneumococcal vaccination.
- Two new pneumococcal vaccines – Prevnar 20 and Vaxneuvance. Med Lett Drugs Ther 2021; 63:188.
- HL Platt et al. Safety, tolerability, and immunogenicity of an adult pneumococcal conjugate vaccine, V116 (STRIDE-3): a randomised, double-blind, active comparator controlled, international phase 3 trial. Lancet Infect Dis 2024 July 1 (epub). doi:10.1016/s1473-3099(24)00344-x
- P Scott et al. A phase 3 clinical study to evaluate the safety, tolerability, and immunogenicity of V116 in pneumococcal vaccine-experienced adults 50 years of age or older (Stride-6). Clin Infect Dis 2024 July 31 (epub). doi:10.1093/cid/ciae383
- American College of Obstetricians and Gynecologists. Maternal immunization. Practice advisory. October 2022. Available at: https://bit.ly/3ys9uyu. Accessed September 26, 2024.
- M Kobayashi et al. Use of 21-valent pneumococcal conjugate vaccine among U.S. adults: recommendations of the Advisory Committee on Immunization Practices - United States, 2024. MMWR Morb Mortal Wkly Rep 2024; 73:793. doi: 10.15585/mmwr.mm7336a3
- Mark Abramowicz, M.D., President has disclosed no relevant financial relationships.
- Jean-Marie Pflomm, Pharm.D., Editor in Chief has disclosed no relevant financial relationships.
- Michael Viscusi, Pharm.D., Associate Editor has disclosed no relevant financial relationships.
- Review the efficacy and safety of inhaled epinephrine (neffy) for emergency treatment of type 1 hypersensitivity reactions including anaphylaxis.
- Description: The first epinephrine nasal spray.
- Indication: Emergency treatment of type 1 hypersensitivity reactions including anaphylaxis in patients who weigh ≥30 kg.
- Clinical Studies: In studies in healthy subjects, the nasal spray appeared to be pharmacologically comparable to injectable epinephrine.
- Adverse Effects: Throat irritation, headache, nasal discomfort, a jittery sensation, tremor, and rhinorrhea can occur.
- Dosage: One 2-mg spray intranasally; the dose can be repeated 5 minutes later if symptoms do not improve.
- Cost: The wholesale acquisition cost of a package containing two single-use devices is $710; according to the manufacturer, the cost for most patients with commercial insurance will not exceed $25.
- Conclusion: The neffy nasal spray could be more convenient to use than injectable epinephrine for some individuals. It has not been studied in persons with underlying structural nasal conditions.
Outline
Table |
The FDA has approved an epinephrine nasal spray (neffy – ARS Pharma) for emergency treatment of type 1 hypersensitivity reactions including anaphylaxis in patients who weigh ≥30 kg. It is the first noninjectable epinephrine product to be approved for this indication.

INJECTABLE EPINEPHRINE — Multiple epinephrine auto-injector formulations are available for emergency treatment of anaphylaxis (see Table 1). EpiPen and its generics have been used effectively for years. A generic version of Adrenaclick (brand no longer manufactured) is similar to EpiPen in size and functionality. AUVI-Q provides visual signals and audio instructions, has an automatic needle retraction system, and appears to be more convenient to carry and easier to use than EpiPen.1 Because of differences in device design and instructions for use, these auto-injectors are not interchangeable and pharmacists cannot substitute one for another.
An epinephrine prefilled syringe (Symjepi) is also FDA-approved for emergency treatment of anaphylaxis. It requires the user to manually inject the needle and push down the plunger, which may be difficult for some patients, particularly children.2
Some injectable epinephrine products have not been consistently available in recent years; at press time, EpiPen, the generic version of Adrenaclick, and Symjepi were on back order with no estimated return date.3
THE NEW FORMULATION — Each neffy nasal spray device contains a single 2-mg dose of epinephrine and is about the size of a teabag; the product is supplied in packages containing two devices. The drug delivery system is the same as that used in several other FDA-approved nasal sprays, including naloxone (Narcan). An absorption-enhancing agent (Intravail), which is also used in nasal spray formulations of drugs such as sumatriptan (Tosymra) and diazepam (Valtoco), increases epinephrine bioavailability.4
CLINICAL STUDIES — As with injectable epinephrine products for treatment of anaphylaxis, no efficacy trials were required for FDA approval of neffy. Approval was based on the results of five pharmacologic studies (summarized in the package insert) in a total of 175 healthy adults and 42 healthy children 8-17 years old who weighed ≥30 kg. Exposure to epinephrine and changes in blood pressure and pulse rate following a 2-mg dose of the nasal spray were comparable to those following a 0.3-mg IM dose, including in subjects with seasonal allergic rhinitis who underwent a nasal allergen challenge. The nasal spray has not been studied in persons with underlying structural nasal conditions, such as polyps or a history of nasal injury or surgery.

ADVERSE EFFECTS — The most common adverse effects of two doses of neffy in adults (incidence 7-19%) were throat irritation, headache, nasal discomfort, a jittery sensation, tremor, and rhinorrhea. Similar adverse effects were observed in children. The nasal spray solution contains sodium metabisulfite, which could cause a hypersensitivity reaction in patients with a sulfite allergy, but a history of sulfite sensitivity should not deter emergency use of the product.
DOSAGE, ADMINISTRATION, AND COST — The recommended dosage of neffy is one 2-mg spray administered into one nostril. If symptoms do not improve after 5 minutes, a second spray may be administered into the same nostril using a new device. The devices should be stored at room temperature, but excursions up to 50° C (122° F) are permitted; injectable epinephrine formulations only permit excursions to 30° C (86° F). At temperatures below -15° C (5° F), the nasal spray solution freezes and the device does not deliver epinephrine. Like other epinephrine products, neffy should be replaced before its expiration date. The shelf life of neffy is 30 months, which is longer than injectable epinephrine products (generally 12-18 months).
According to the manufacturer, the out-of-pocket cost for two neffy nasal spray devices should not exceed $25 for most patients with commercial insurance or $199 for those using coupons from discount sites such as GoodRx.5
CONCLUSION — The neffy nasal spray offers a new, potentially more convenient route of epinephrine delivery for emergency treatment of anaphylaxis in adults and children who weigh ≥30 kg. Whether its effectiveness differs from that of injectable epinephrine remains to be determined. Until data on the efficacy of neffy become available, some expert clinicians are advising patients to also carry an injectable epinephrine product. Neffy has not been studied in patients with underlying structural nasal conditions, such as polyps or a history of injury or surgery.
- Auvi-Q epinephrine auto-injector returns. Med Lett Drugs Ther 2017; 59:33.
- An epinephrine prefilled syringe (Symjepi) for anaphylaxis. Med Lett Drugs Ther 2019; 61:25.
- American Society of Health-System Pharmacists. Current drug shortages. Available at: https://bit.ly/3Qs7ETT. Accessed September 26, 2024.
- AK Ellis et al. Development of neffy, an epinephrine nasal spray, for severe allergic reactions. Pharmaceutics 2024; 16:811. doi:10.3390/pharmaceutics16060811
- ARS Pharma Press Release. ARS Pharmaceuticals receives FDA approval of neffy (epinephrine nasal spray), the first and only needle-free treatment for type I allergic reactions, including anaphylaxis. August 9, 2024. Available at: https://bit.ly/4cyySk4. Accessed September 26, 2024.
- Mark Abramowicz, M.D., President has disclosed no relevant financial relationships.
- Jean-Marie Pflomm, Pharm.D., Editor in Chief has disclosed no relevant financial relationships.
- Brinda M. Shah, Pharm.D., Consulting Editor has disclosed no relevant financial relationships.
- Review the efficacy and safety of vonoprazan (Voquezna) for treatment of heartburn associated with nonerosive gastroesophageal reflux disease (GERD).
- Description: Oral potassium-competitive acid blocker.
- Indication: Relief of heartburn associated with nonerosive GERD in adults.
- Efficacy: Vonoprazan was associated with more heartburn-free days compared to placebo in one 4-week trial.
- Adverse Effects: GI adverse effects and urinary tract infection were most common.
- Drug Interactions: Vonoprazan may interact with drugs that induce CYP3A4, are substrates of CYP2C19, or require gastric acid for absorption.
- Dosage: 10 mg orally once daily.
- Cost: A 28-day supply costs $606.
- Conclusion: Until more long-term safety data become available, other acid-suppressing drugs are preferred.
| Outline |
The potassium-competitive acid blocker vonoprazan (Voquezna – Phathom), which was approved earlier for treatment of erosive esophagitis, has now been approved by the FDA for relief of heartburn associated with nonerosive gastroesophageal reflux disease (GERD) in adults.1 Vonoprazan is also available copackaged with amoxicillin (Voquezna Dual Pak) and with amoxicillin and clarithromycin (Voquezna Triple Pak) for treatment of Helicobacter pylori infection.2

GERD ― Heartburn and regurgitation are the classic symptoms of GERD. Other symptoms may include chest pain and chronic cough. Drugs that suppress gastric acid are the standard treatment for GERD. Proton pump inhibitors (PPIs) are more effective than H2-receptor antagonists (H2RAs) in relieving heartburn. Most patients will have a relapse of symptoms after stopping treatment; long-term maintenance treatment is often necessary.3,4
PHARMACOLOGY ― Vonoprazan blocks the final step of acid secretion in the gastric parietal cell by competitively blocking potassium binding to hydrogen-potassium ATPase (the "proton pump"). Unlike PPIs, vonoprazan does not require activation by acid. Vonoprazan is more rapidly absorbed, has a longer half-life (~7 hours vs ~1-2 hours), and achieves a higher intragastric pH than PPIs.5
CLINICAL STUDIES ― FDA approval of vonoprazan for the new indication was based on the results of a double-blind trial in 772 patients with nonerosive GERD who had heartburn on at least 4 of 7 days prior to randomization. Patients were randomized to receive vonoprazan 10 or 20 mg or placebo once daily for 4 weeks. After 4 weeks, patients taking placebo were rerandomized to receive vonoprazan 10 or 20 mg for an additional 20 weeks; those already taking vonoprazan continued taking their original dose. The percentage of heartburn-free days through week 4, the primary endpoint, was statistically significantly greater with both doses of vonoprazan than with placebo (44.8% with 10 mg and 44.4% with 20 mg vs 27.7%). The mean percentage of heartburnfree days during the 20-week extension period with vonoprazan was 61-63%.6
In a trial in 483 patients in Japan with nonerosive GERD who were randomized to receive vonoprazan 10 mg or placebo once daily for 4 weeks, the percentage of heartburn-free days, the primary endpoint, was not significantly greater with vonoprazan compared to placebo (72.6% vs 61.5%).7
No trials directly comparing vonoprazan to PPIs or H2RAs for treatment of nonerosive GERD are available.
ADVERSE EFFECTS ― In the pivotal clinical trial, the most common adverse effects (frequency ≥2%) of vonoprazan were abdominal pain, constipation, diarrhea, nausea, and urinary tract infection. As with PPIs, Clostridioides difficile infection, acute tubulointerstitial nephritis, bone fractures, and severe cutaneous adverse reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis can occur with use of vonoprazan. Vitamin B12 deficiency and hypomagnesemia have also been reported. Hyperplasia of the gastric mucosa and gastric hyperplastic polyps with chronic bleeding have been reported with longterm use of vonoprazan in patients with erosive esophagitis.8
DRUG INTERACTIONS ― Vonoprazan can decrease serum concentrations of drugs that require gastric acidity for absorption, such as the antiretroviral drugs rilpivirine and atazanavir. Vonoprazan is a CYP3A4 substrate; use with strong or moderate CYP3A4 inducers can decrease serum concentrations of vonoprazan and should be avoided. Vonoprazan is an inhibitor of CYP2C19; it may increase serum concentrations of CYP2C19 substrates and can reduce the antiplatelet effect of clopidogrel, which is converted to its active form by CYP2C19.9
PREGNANCY AND LACTATION ― No adequate data are available on vonoprazan use in pregnant women. In animal studies, liver lesions and increased stomach weight were reported in rats that had gestational or lactational exposure to vonoprazan. Vonoprazan and its metabolites are present in rat milk. No data are available on the presence of vonoprazan in breast milk or its effects on the breastfed infant or milk production. Breastfeeding is not recommended during treatment with the drug.
DOSAGE, ADMINISTRATION, AND COST ― Voquezna is supplied in 10- and 20-mg tablets. The recommended dosage for relief of heartburn associated with nonerosive GERD is 10 mg once daily for 4 weeks. The tablets should be swallowed whole; they should not be chewed or crushed. The wholesale acquisition cost (WAC) for a 28-day supply of Voquezna is $606.10
CONCLUSION ― The potassium-competitive acid blocker vonoprazan (Voquezna) reduced heartburn significantly in patients with nonerosive GERD in one 4-week placebo-controlled trial, but not in a second trial. Data on its long-term safety are lacking. Older, less expensive acid-suppressing drugs should be tried first.
- Vonoprazan (Voquezna) for erosive esophagitis. Med Lett Drugs Ther 2023; 65:203.
- Two vonoprazan combinations (Voquezna) for H. pylori. Med Lett Drugs Ther 2022; 64:169.
- PO Katz et al. ACG clinical guideline for the diagnosis and management of gastroesophageal reflux disease. Am J Gastroenterol 2022; 117:27. doi:10.14309/ajg.0000000000001538
- Drugs for GERD and peptic ulcer disease. Med Lett Drugs Ther 2022; 64:49.
- DY Graham and MP Dore. Update on the use of vonoprazan: a competitive acid blocker. Gastroenterology 2018; 154:462. doi:10.1053/j.gastro.2018.01.018
- L Laine et al. Vonoprazan is efficacious for treatment of heartburn in non-erosive reflux disease: a randomized trial. Clin Gastroenterol Hepatol 2024 May 14 (epub). doi:10.1016/j.cgh.2024.05.004
- Y Kinoshita et al. Efficacy and safety of vonoprazan in patients with nonerosive gastroesophageal reflux disease: a randomized, placebo-controlled, phase 3 study. Clin Transl Gastroenterol 2019; 10:e00101. doi:10.14309/ctg.0000000000000101
- C Goto et al. Long-term vonoprazan administration causes gastric fundic gland-type hyperplastic polyps and chronic bleeding. Clin J Gastroenterol 2023; 16:159. doi:10.1007/s12328-022-01751-0
- Inhibitors and inducers of CYP enzymes and P-glycoprotein and other transporters. Med Lett Drugs Ther 2023 January 25 (epub). Available at: www.medicalletter.org/downloads/CYP_PGP_Tables.pdf.
- Approximate WAC. WAC = wholesaler acquisition cost or manufacturer’s published price to wholesalers; WAC represents a published catalogue or list price and may not represent an actual transactional price. Source: AnalySource® Monthly. August 5, 2024. Reprinted with permission by First Databank, Inc. All rights reserved. ©2024. www.fdbhealth.com/policies/drug-pricing-policy.
- Mark Abramowicz, M.D., President has disclosed no relevant financial relationships.
- Jean-Marie Pflomm, Pharm.D., Editor in Chief has disclosed no relevant financial relationships.
- Amy Faucard, MLS, Associate Editor has disclosed no relevant financial relationships.
- Review the efficacy and safety of mResvia, an mRNA respiratory syncytial virus (RSV) vaccine, for prevention of lower respiratory tract disease caused by RSV in adults ≥60 years old.
- Description: An mRNA respiratory syncytial virus (RSV) vaccine encoding a stabilized prefusion form of the RSV fusion (F) glycoprotein.
- Indication: FDA-approved for prevention of lower respiratory tract disease (LRTD) caused by RSV in adults ≥60 years old.
- Efficacy: A single dose has prevented RSV LRTD in adults ≥60 years old compared to placebo. One dose of mResvia appears to be less effective over two RSV seasons than one dose of a recombinant RSV vaccine (Arexvy or Abrysvo).
- Adverse Effects: Most common are injection-site pain, axillary swelling or tenderness, fatigue, headache, myalgia, arthralgia, and chills.
- Dosage: A single 50 mcg/0.5 mL IM dose.
- Cost: The wholesale acquisition cost for one dose is $290.
- Conclusion: The CDC Advisory Committee on Immunization Practices (ACIP) recommends a single dose of any available RSV vaccine for all adults ≥75 years old and for those 60-74 years old at increased risk of severe RSV disease.
Outline
Tables |
The FDA has licensed mResvia (Moderna), an mRNA respiratory syncytial virus (RSV) vaccine, for prevention of lower respiratory tract disease (LRTD) caused by RSV in adults ≥60 years old. It is the first mRNA vaccine to be licensed in the US for this indication. Two recombinant RSV vaccines, Arexvy and Abrysvo, are also available for prevention of RSV LRTD.1 Arexvy is approved for use in adults ≥50 years old.2 Abrysvo is approved for use in adults ≥60 years old and in pregnant women to prevent RSV LRTD in their infants.
RSV DISEASE — RSV typically causes a mild upper respiratory tract infection in adults, but older adults, particularly those with underlying health conditions, have an increased risk of RSV-associated hospitalization. RSV epidemics in the Northern Hemisphere typically occur between October and April, peaking in December or January.
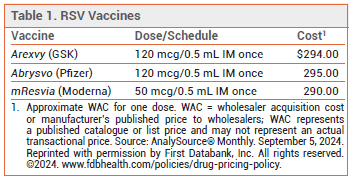
EFFICACY OF RECOMBINANT VACCINES — Both Arexvy and Abrysvo have been shown to reduce the incidence of RSV LRTD in adults ≥60 years old.1 A case-control analysis of patients ≥60 years old hospitalized for acute respiratory illness found that vaccination with a recombinant RSV vaccine was about 75% effective against RSV-associated hospitalization during the first season of use.3 Use of Abrysvo in pregnant women reduced the incidence of medically-attended RSV LRTD in their infants during one RSV season.1
THE NEW VACCINE — mResvia is a lipid nanoparticle-encapsulated, mRNA-based vaccine. It encodes a stabilized prefusion form of the RSV fusion (F) glycoprotein derived from an RSV A strain. The RSV prefusion F glycoprotein mediates viral fusion and host-cell entry and elicits neutralizing antibodies. The same antigen is used in Arexvy and Abrysvo.

CLINICAL STUDIES — FDA approval of mResvia was based on the preliminary results of an ongoing observer-blinded trial (ConquerRSV) in 35,064 adults ≥60 years old who were randomized to receive a single dose of mResvia or placebo. The vaccine was effective, compared to placebo, in preventing a first episode of RSV LRTD (see Table 2). Vaccine efficacy against RSV-associated acute respiratory disease was 68.4%; efficacy against RSV A was higher than that against RSV B (78.5% vs 51.7%).4
A follow-up report found that a single dose of mResvia was ~50% effective in preventing RSV LRTD over 18 months in adults ≥60 years old.5 Higher efficacy rates against RSV LRTD over two RSV seasons have been reported following a single dose of Arexvy or Abrysvo (67.7% over 23.3 months with Arexvy and 81.5% over 16.4 months with Abrysvo).6,7
ADVERSE EFFECTS — In the ConquerRSV trial, the most common local adverse effects reported within 7 days of mResvia vaccination were injection-site pain (55.9%) and axillary swelling or tenderness (15.2%). Systemic adverse effects included fatigue (30.8%), headache (26.7%), myalgia (25.6%), arthralgia (21.7%), and chills (11.6%). More cases of urticaria were reported with mResvia than with placebo within 28 days after administration (15 vs 5). One participant in the mResvia group developed facial paralysis 4 days after vaccination. Atrial fibrillation and Guillain-Barré syndrome (GBS) were reported following administration of Arexvy or Abrysvo in clinical trials. Vaccine-related cardiac arrhythmias and GBS were not reported in clinical trials with mResvia.5

ACIP RECOMMENDATIONS — The CDC Advisory Committee on Immunization Practices (ACIP) has updated its recommendations for use of RSV vaccines in older adults. A single dose of an RSV vaccine is now recommended for all adults ≥75 years old and for those 60-74 years old at increased risk of severe RSV disease (see Table 3).8 For optimal protection, the vaccine should be given before the onset of the RSV season. Administration of an RSV vaccine and other adult vaccines during the same visit is considered acceptable, but local or systemic reactogenicity could increase.9
DOSAGE AND ADMINISTRATION — The mResvia vaccine is supplied as a frozen suspension in prefilled syringes containing a single 50 mcg/0.5 mL IM dose. The syringe can be thawed in the refrigerator for 60 minutes and should stand at room temperature for 10-20 minutes before administration. A syringe that is thawed at room temperature is ready to be administered after 45 minutes.
CONCLUSION — A single dose of mResvia, the first mRNA respiratory syncytial virus (RSV) vaccine, was effective in preventing RSV-associated lower respiratory tract disease during one RSV season in adults ≥60 years old. Follow-up data suggest that one dose of mResvia may offer less protection over two RSV seasons than one dose of Arexvy or Abrysvo, the recombinant RSV vaccines available in the US. The CDC Advisory Committee on Immunization Practices recommends a single dose of any available RSV vaccine for all adults ≥75 years old and for those 60-74 years old at increased risk of severe RSV disease.
- Two vaccines (Arexvy and Abrysvo) for prevention of RSV disease. Med Lett Drugs Ther 2023; 65:155.
- In brief: RSV vaccine (Arexvy) for ages 50-59. Med Lett Drugs Ther 2024; 66:113.
- D Surie et al. RSV vaccine effectiveness against hospitalization among US adults 60 years and older. JAMA 2024 Sep 4 (epub). doi:10.1001/jama.2024.15775
- E Wilson et al. Efficacy and safety of an mRNA-based RSV preF vaccine in older adults. N Engl J Med 2023; 389:2233. doi:10.1056/nejmoa2307079
- R Das. Update on Moderna's RSV vaccine, mResvia (mRNA-1345), in adults ≥60 years of age. ACIP presentation slides: June 26-28, 2024 meeting. Available at: https://bit. ly/4ciO7hH. Accessed September 26, 2024.
- I Munjal. RSVpreF adult clinical development update. ACIP presentation slides: June 26-28, 2024 meeting. Available at: https://bit.ly/4ciO7hH. Accessed September 26, 2024.
- S Gerber. Arexvy (adjuvanted RSVPreF3) 2-year update. ACIP presentation slides: June 26-28, 2024 meeting. Available at: https://bit.ly/4ciO7hH. Accessed September 26, 2024.
- A Britton et al. Use of respiratory syncytial virus vaccines in adults aged ≥60 years: updated recommendations of the Advisory Committee on Immunization Practices – United States, 2024. MMWR Morb Mortal Wkly Rep 2024; 73:696. doi:10.15585/mmwr.mm7332e1
- CDC. Vaccines and immunizations. Healthcare providers: RSV vaccination for adults 60 years of age and over. July 3, 2024. Available at: https://bit.ly/3yC0A1n. Accessed September 26, 2024.
The FDA has required a new warning in the label of the oral selective neurokinin 3 (NK3) receptor antagonist fezolinetant (Veozah) about the risk of hepatoxicity.1,2 The label of fezolinetant, which was approved by the FDA in 2023 for treatment of moderate to severe vasomotor symptoms due to menopause, already contained a warning about hepatic transaminase elevations associated with use of the drug.
The new warning was based on a single post-marketing case of acute mixed hepatocellular cholestatic drug-induced liver injury. The patient presented with fatigue, nausea, itching, jaundice, light-colored stools, dark urine, and elevated hepatic enzyme and total bilirubin levels within 40 days of starting fezolinetant.
Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), and bilirubin levels should be checked before starting fezolinetant, monthly for the first 3 months, and at 6 and 9 months after starting the drug. Fezolinetant should not be started if ALT or AST concentrations are ≥2 times the upper limit of normal (ULN) or if total bilirubin is elevated. The drug should be stopped if transaminase levels are >5 times the ULN or if transaminase levels are >3 times the ULN and total bilirubin level is >2 times the ULN.
- FDA Drug Safety Communication. FDA adds warning about rare occurrence of serious liver injury with use of Veozah (fezolinetant) for hot flashes due to menopause. Stop medicine if signs and symptoms of liver injury occur. September 12, 2024. Available at: https://bit.ly/4ghd4MK. Accessed September 26, 2024.
- Fezolinetant (Veozah) for menopausal vasomotor symptoms. Med Lett Drugs Ther 2023; 65:97.
- Mark Abramowicz, M.D., President has disclosed no relevant financial relationships.
- Jean-Marie Pflomm, Pharm.D., Editor in Chief has disclosed no relevant financial relationships.
- Review the efficacy and safety of Capvaxive, a 21-valent pneumococcal conjugate vaccine, for prevention of invasive pneumococcal disease and pneumococcal pneumonia in adults.
View Figure 1: Pneumococcal Vaccine Recommendations for Adults 19-64 Years Old
Download PDF: US English

- Mark Abramowicz, M.D., President has disclosed no relevant financial relationships.
- Jean-Marie Pflomm, Pharm.D., Editor in Chief has disclosed no relevant financial relationships.
- Review the efficacy and safety of Capvaxive, a 21-valent pneumococcal conjugate vaccine, for prevention of invasive pneumococcal disease and pneumococcal pneumonia in adults.
View Figure 2: Pneumococcal Vaccine Recommendations for Adults ≥65 Years Old
Download PDF: US English

A reader of our article on the 2024-2025 formulations of the mRNA COVID-19 vaccines manufactured by Pfizer/BioNTech (Comirnaty) and Moderna (Spikevax)1 asked us to provide more information on the data that supported their licensure.
The vaccines were initially licensed by the FDA in 2021 (Pfizer) and 2022 (Moderna) based on the results of large, double-blind trials in SARS-CoV-2-naive adolescents and adults. Both vaccines significantly decreased the risk of symptomatic and severe SARS-CoV-2 infection compared to placebo.2-4 Issuance of the FDA Emergency Use Authorizations (EUAs) allowing use of the vaccines in younger children was initially based primarily on the results of immunogenicity studies. Titer levels of anti-SARS-CoV-2 neutralizing antibodies following vaccination were at least as high in persons 6 months through 11 years old as they were in comparator adult cohorts.5,6
As with the influenza vaccine, efficacy trials are no longer required by the FDA for licensure of new COVID-19 vaccine formulations that only significantly differ from previous formulations in the virus strain that they target. Licensure is based on the immunogenicity, efficacy, and safety of previous formulations, and on the likelihood of the new formulations to protect against currently circulating SARS-CoV-2 variants.7-9
Observational studies suggest that all of the COVID-19 vaccine formulations available in the US in 2023-2024 were effective in reducing the incidence of COVID-19. In a case-control analysis of 14,860 COVID-19 nucleic acid amplification tests administered at community pharmacies to immunocompetent adults with COVID-like symptoms between September 2023 and May 2024, receipt of any 2023-2024 COVID-19 vaccine at least 7 days before the test was associated with a decreased incidence of SARS-CoV-2 infection. The estimated adjusted vaccine efficacy was 45%; it was 58% for infections likely caused by the XBB.1.5 variant, which the 2023-2024 vaccines targeted, and 37% for infections likely caused by the JN.1 variant.10,11
In similar analyses of tests administered to adults with COVID-like illness within 10 days before or 3 days after an emergency department/urgent care visit (n=245,504) or a hospitalization (n=77,103) between September 2023 and May 2024, receipt of any 2023-2024 COVID-19 vaccine at least 7 days before the test was associated with a decreased incidence of COVID-19 requiring an emergency department/urgent care visit (estimated adjusted vaccine efficacy 36%) or hospitalization (estimated adjusted vaccine efficacy 41%). Among hospitalized immunocompetent persons, the estimated adjusted vaccine efficacy against critical illness was 58%.10,12
- COVID-19 update: New Pfizer and Moderna vaccine formulations for 2024-2025. Med Lett Drugs Ther 2024; 66:151.
- FP Polack et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N Engl J Med 2020; 383:2603. doi:10.1056/nejmoa2034577
- LR Baden et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med 2021; 384:403. doi:10.1056/nejmoa2035389
- K Ali et al. Evaluation of mRNA-1273 SARS-CoV-2 vaccine in adolescents. N Engl J Med 2021; 385:2241. doi:10.1056/nejmoa2109522
- FDA. Fact sheet for healthcare providers administering vaccine: Emergency Use Authorization (EUA) of Pfizer-BioNTech COVID-19 vaccine (2024-2025 formula), for 6 months through 11 years of age. August 22, 2024. Available at: https://bit.ly/4g2dQxa. Accessed September 26, 2024.
- FDA. Fact sheet for healthcare providers administering vaccine: Emergency Use Authorization (EUA) of Moderna COVID-19 vaccine (2024-2025 formula), for individuals 6 months through 11 years of age. August 2024. Available at: https://bit.ly/3YZmmqo. Accessed September 26, 2024.
- FDA News Release. FDA approves and authorizes updated mRNA COVID-19 vaccines to better protect against currently circulating variants. August 22, 2024. Available at: https://bit.ly/3T3eGzP. Accessed September 26, 2024.
- FDA CBER. Summary minutes. 185th Vaccines and Related Biological Products Advisory Committee. June 5, 2024 Available at: https://bit.ly/4enFzqm. Accessed September 26, 2024.
- COVID-19 Real-Time Learning Network. COVID-19 variant update. September 17, 2024. Available at: https://bit.ly/3XslucR. Accessed September 26, 2024.
- R Link-Gelles. Effectiveness of COVID-19 (2023-2024 formula) vaccines. ACIP Meeting COVID-19 Vaccines. June 27, 2024. Available at: https://bit.ly/4gA1ixl. Accessed September 26, 2024.
- R Link-Gelles et al. Early estimates of updated 2023-2024 (monovalent XBB.1.5) COVID-19 vaccine effectiveness against symptomatic SARS-CoV-2 infection attributable to co-circulating Omicron variants among immunocompetent adults - increasing community access to testing program, United States, September 2023-January 2024. MMWR Morb Mortal Wkly Rep 2024; 73:77. doi:10.15585/mmwr.mm7304a2
- J DeCuir et al. Interim effectiveness of updated 2023–2024 (monovalent XBB.1.5) COVID-19 vaccines against COVID-19–associated emergency department and urgent care encounters and hospitalization among immunocompetent adults aged ≥18 Years — VISION and IVY Networks, September 2023–January 2024. MMWR Morb Mortal Wkly Rep 2024; 73:180. doi:10.15585/mmwr.mm7308a5
- Description: A melanoma-associated antigen A4 (MAGEA-4)- directed genetically modified autologous T-cell immunotherapy.
- Indication: Treatment of adults with unresectable or metastatic synovial sarcoma who received prior chemotherapy and are HLA-A*02:01P, -A*02:02P, -A*02:03P, or -A*02:06P positive and whose tumor expresses the MAGE-A4 antigen.
- Efficacy: A single infusion of the immunotherapy produced an overall response rate of 39%.
- Adverse Effects: Cytokine release syndrome, immune effector cell-associated neurotoxicity, prolonged severe cytopenias, severe infection, and secondary malignancies can occur.
- Dosage: A single IV infusion of 2.68 x 109 to 10 x 109 MAGE-A4 TCR positive T cells.
- Cost: One dose costs $727,000.
- Conclusion: About 40% of patients with previously treated unresectable or metastatic synovial sarcoma who received a single infusion of Tecelra achieved a response.
| Outline |
Afamitresgene autoleucel (Tecelra – Adaptimmune), a melanoma-associated antigen A4 (MAGE-A4)-directed genetically modified autologous T-cell immunotherapy, has received accelerated approval from the FDA for one-time treatment of adults with unresectable or metastatic synovial sarcoma who received prior chemotherapy and are HLA-A*02:01P, -A*02:02P, -A*02:03P, or -A*02:06P positive and whose tumor expresses the MAGE-A4 antigen. It is the first gene therapy to be approved in the US for treatment of synovial sarcoma. Accelerated approval of the immunotherapy was based on the overall response rate and duration of response.

THE PRODUCT ― Tecelra is prepared from autologous peripheral blood mononuclear cells obtained by leukapheresis. The cells are sent to a commercial laboratory where they are enriched for T cells and then transduced with a replication-incompetent lentiviral vector containing the MAGE-A4 TCR transgene. Tecelra is formulated as a cell suspension containing 2.68 x 109 to 10 x 109 MAGE-A4 TCR positive T cells. Tecelra contains dimethyl sulfoxide (DMSO), which can cause serious hypersensitivity reactions.
MECHANISM OF ACTION ― MAGE-A4 peptide is an intracellular cancer-testis antigen that is expressed in synovial sarcoma. The autologous T-cell immunotherapy recognizes an HLA-A*02 restricted MAGE-A4 peptide expressed on the cell surface. Activation of Tecelra results in T cell proliferation, cytokine secretion, and death of MAGE-A4/HLA-A* 02-expressing synovial sarcoma cells.
CLINICAL STUDIES ― FDA approval of afamitresgene autoleucel was based on the results of an open-label trial (SPEARHEAD-1) in 44 HLA-A*02:01-03 and 06 allele-positive patients with inoperable or metastatic synovial sarcoma who received prior systemic chemotherapy (doxorubicin and/or ifosfamide) and whose tumor expressed the MAGE-A4 antigen. All patients received a single infusion of afamitresgene autoleucel. After a median follow-up of 32.6 months, the overall response rate was 39%.1
ADVERSE EFFECTS ― The Tecelra label includes a boxed warning about the risk of cytokine release syndrome (CRS), which can cause hypotension, pulmonary edema, coagulopathy, multiorgan failure, and death. Nausea, vomiting, fatigue, pyrexia, constipation, dyspnea, abdominal pain, decreased appetite, tachycardia, back pain, hypotension, diarrhea, and edema can occur. Immune effector cell-associated neurotoxicity syndrome (ICANS), prolonged severe cytopenias, severe infection, and secondary malignancies have been reported. Patients should avoid driving or engaging in hazardous occupations or activities for at least 4 weeks after receiving Tecelra. The immunotherapy is contraindicated in patients who are heterozygous or homozygous for HLA-A*02:05P.
DOSAGE, ADMINISTRATION, AND COST ― Patients should receive lymphodepleting chemotherapy (cyclophosphamide and fludarabine) starting the seventh day before the Tecelra infusion. Acetaminophen and an H1-antihistamine should be given about 30-60 minutes before the Tecelra infusion. Administration of a granulocyte-colony stimulating factor (G-CSF) can be considered for patients with neutropenia. Patients should be monitored at the treatment facility for at least 7 days after receiving Tecelra and stay near the treatment facility for at least 4 weeks. One dose of Tecelra costs $727,000.2
- SP D’Angelo et al. Afamitresgene autoleucel for advanced synovial sarcoma and myxoid round cell liposarcoma (SPEARHEAD-1): an international, open-label, phase 2 trial. Lancet 2024; 403:1460. doi:10.1016/S0140-6736(24)00319-2
- Approximate WAC. WAC = wholesaler acquisition cost or manufacturer’s published price to wholesalers; WAC represents a published catalogue or list price and may not represent an actual transactional price. Source: AnalySource® Monthly. September 5, 2024. Reprinted with permission by First Databank, Inc. All rights reserved. ©2023. www.fdbhealth.com/policies/drug-pricing-policy.
The FDA has approved Erzofri (Luye), an extended-release injectable formulation of the second-generation antipsychotic drug paliperidone palmitate, for treatment of schizophrenia and schizoaffective disorder in adults. It is the second once-monthly formulation of paliperidone palmitate to be approved in the US for these indications; Invega Sustenna was the first. Longer-acting injectable formulations of paliperidone palmitate are also available (see Table 1).1

LONG-ACTING INJECTABLE ANTIPSYCHOTICS ― Extended-release injectable second-generation antipsychotic drugs are generally used in patients with a history of relapse due to poor adherence to oral maintenance therapy.2
CLINICAL STUDIES ― No new clinical efficacy trials were required for FDA approval of Erzofri; approval was based on earlier trials with Invega Sustenna and an open-label study showing that the bioavailability of paliperidone palmitate with Erzofri was similar to that with Invega Sustenna.3
DOSAGE AND ADMINISTRATION ― The initial dose of Erzofri is 351 mg injected IM into the deltoid muscle; subsequent doses can be given in the gluteal or deltoid muscle. For schizophrenia, the recommended dosage is 117 mg IM (range 39-234 mg) once monthly. In patients with mild renal impairment, the recommended initial dose is 234 mg, followed by 78 mg once monthly (max 156 mg). Erzofri is not recommended for use in patients with moderate or severe renal impairment.
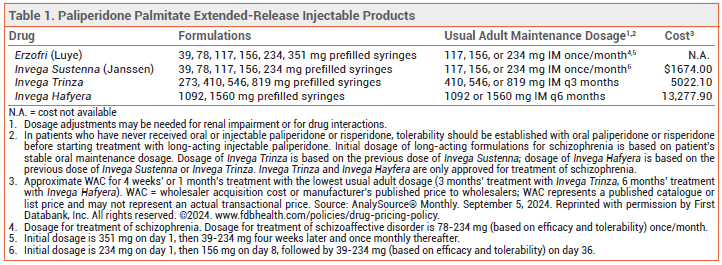
CONCLUSION ― A once-monthly, extended-release injectable formulation of the second-generation antipsychotic drug paliperidone palmitate (Invega Sustenna) has been available for years. Erzofri has not been shown to offer any advantage in efficacy or safety over Invega Sustenna. Extended-release formulations of paliperidone palmitate that are administered once every 3 months or 6 months are also available and may be preferred.
- In brief: Twice-yearly paliperidone (Invega Hafyera) for schizophrenia. Med Lett Drugs Ther 2022; 64:7.
- GA Keepers et al. The American Psychiatric Association practice guideline for the treatment of patients with schizophrenia. Am J Psychiatry 2020; 177:868. doi:10.1176/appi.ajp.2020.177901
- ClinicalKey Clinical Trial. A study to evaluate the PK profiles of LY03010 and relative bioavailability at steady-state of LY03010 versus Invega Sustenna in schizophrenia patients. Available at: https://bit.ly/4gc2jLQ. Accessed September 12, 2024.
The Medical Letter, Inc. does not warrant that all the material in this publication is accurate and complete in every respect. The Medical Letter, Inc. and its editors shall not be held responsible for any damage resulting from any error, inaccuracy, or omission.